
 UK (English)
UK (English) Deutsch
Deutsch عربى
عربى Français
Français Hrvatska
Hrvatska Русский
Русский Türkçe
Türkçe
Educație

Ce este boala CLN2?
CLN2 este o boală genetică
Boala CLN2 este o tulburare genetică rară care afectează copiii.1,2 Boala CLN2 este:
- Numită după gena CLN2/TPP1, care este mutantă (anormală) în boala CLN22
- Una din cele mai frecvente forme de lipofuscinoză ceroidă neuronală (NCL)2
- NCL sunt un grup de tulburări ereditare, cunoscute colectiv ca boala Batten3
- Cunoscută anterior ca NCL infantilă tardivă, pentru că la majoritatea copiilor simptomele încep între vârstele de 2 și 4 ani2
Boala CLN2 este o boală ereditară care este moștenită în familie
Copiii cu boala CLN2 sunt născuți cu această afecțiune, chiar dacă pot trece luni sau ani înainte ca ei să înceapă să arate semne.2
Boala CLN2 este considerată o tulburare autozomală recesivă2
- Fiecare individ are 2 copii ale genei CLN2. La cei cu boala CLN2, ambele gene moștenite (câte una de la fiecare părinte) au mutații2
- Părinții unui copil cu boala CLN2 au o mutație a uneia din genele lor CLN22
- Părinții sunt purtători ai mutației genetice, ceea ce înseamnă că sunt sănătoși, dar pot transmite mai departe mutația la copiii lor2
- Daca ambii părinții poartă mutația și au un copil, există2
- O probabilitate de 25% ca acesta să moștenească ambele mutații și să fie afectat de boala CLN2, având simptome
- O probabilitate de 50% să fie sănătos, dar va purta și el o mutație pentru boala CLN2
- O probabilitate de 25% să nu poarte nicio mutație și să nu aibă boala CLN2
Cum este diagnosticată CLN2?
Diagnosticul poate fi confirmat prin testare genetică, căutând o mutație într-o probă de ADN recoltată de la copil. Diagnosticul de CLN2 poate fi confirmat și prin testarea enzimatică, în care se caută niveluri scăzute ale enzimei TPP1.4
Boala CLN2 este un tip de tulburare a depozitelor lizozomale, care afectează celulele din creier5
Există lizozomi în fiecare celulă. Lizozomii conțin enzime care descompun și reciclează material din celulă. Una din aceste enzime este denumită TPP1.5
Enzima TPP1 lipsește sau nu funcționează normal la copiii cu boala CLN2. Atunci când enzima nu funcționează corect, anumite substanțe se acumulează în lizozomii celulelor, în special în celulele din creier și ochi.3,6
Boala CLN2 este asociată cu acumularea unor substanțe în interiorul celulelor creierului3,6

CLN2 disease lysosomal storage disorder cell graphic
În timp, această acumulare se asociază cu lezarea celulelor din creier și ochi, care nu mai funcționează normal. Pe măsură ce se întâmplă acest lucru, apar simptomele bolii CLN2 (de exemplu, întârzierea dezvoltării limbajului, convulsiile și tulburările de vedere).1,3
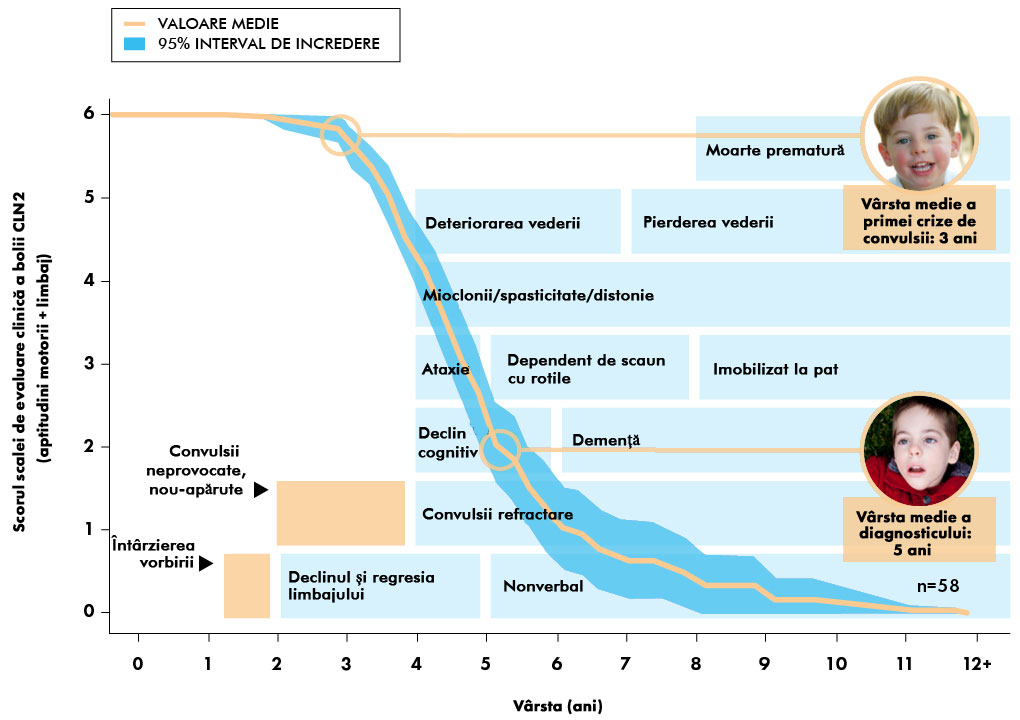
Când apar de obicei simptomele bolii CLN2?
Simptomele bolii CLN2 se înrăutățesc de obicei de-a lungul timpului și includ următoarele:2,5,7-11
BDSRA logo
Surse principale
- Kohlschütter A, Schulz A. CLN2 disease (classic late infantile neuronal ceroid lipofuscinosis) (lipofuscinoza ceroidă neuronală infantilă tardivă clasică). Pediatr Endocrinol Rev. 2016;13(Suppl 1):682-688.
- Mole SE, Williams RE. Neuronal ceroid-lipofuscinoses (lipofuscinozele ceroide neuronale). 2001 Oct 10 [Actualizat la 1 aug. 2013]. In: Pagon RA, et al., eds. GeneReviews®.
- Haltia M. The neuronal ceroid-lipofuscinoses: from past to present (Lipofuscinozele ceroide neuronale: din trecut până în prezent) BiochimBiophys Acta. 2006;1762:850-856.
- Fietz M et al. Diagnosis of neuronal ceroid lipofuscinosis type 2 (CLN2 disease): Expert recommendations for early detection and laboratory diagnosis. (Diagnosticul lipofuscinozei ceroide neuronale de tip 2 (boala CLN2): recomandările experților pentru detectarea timpurie și diagnosticul de laborator). Mol Genet Metab. 2016;119:160-167.
- Mole SE et al. Correlations between genotype, ultrastructural morphology and clinical phenotype in the neuronal ceroid lipofuscinoses. Neurogenetics. (Corelații între genotipul, morfologia ultrastructurală și fenotipul clinic în lipofuscinozele ceroide neuronale. Neurgenetică) 2005;6:107-126.
- Schulz A et al. NCL diseases–clinical perspectives. (Perspectivele clinice ale bolilor NCL) BiochimBiophys Acta. 2013;1832:1801-1806.
- Chang M et al. CLN2. In: Mole S, Williams R, and Goebel H, eds. The neuronal ceroid lipofuscinoses (Batten Disease). (Lipofuscinozele ceroide neuronale (Boala Batten). A 2-a ediție. Oxford, Regatul Unit: Oxford University Press; 2011:80-109.
- Pérez-Poyato MS et al. Late infantile neuronal ceroid lipofuscinosis: mutations in the CLN2 gene and clinical course in Spanish patients. (Lipofuscinoza ceroidă neuronală infantilă tardivă: mutațiile genei CLN2 și evoluția clinică la pacienți spanioli) J Child Neurol. 2013;28:470-478.
- Worgall S et al. Treatment of late infantile neuronal ceroid lipofuscinosis by CNS administration of a serotype 2 adeno-associated virus expressing CLN2 cDNA. (Tratamentul lipofuscinozei ceroide neuronale infantile tardive prin administrarea unui serotip 2 viral asociat adenovirusului, care exprimă CLN2 cADN) Hum Gene Ther. 2008;19:463-474.
- Steinfeld R et al. Late infantile neuronal ceroid lipofuscinosis: quantitative description of the clinical course in patients with CLN2 mutations. (Lipofuscinoza ceroidă neuronală infantilă tardivă : descriere cantitativă a evoluției clinice la pacienții cu mutații CLN2) Am J Med Genet. 2002;112:347-354.
- Nickel M et al. Disease characteristics and progression in patients with late-infantile neuronal ceroid lipofuscinosis type 2 (CLN2) disease: an observational cohort study. (Caracteristicile și evoluția bolii la pacienții cu lipofuscinoză ceroidă neuronală infantilă tardivă tip 2 (boala CLN2): un studiu de cohort observațional) Lancet Child Adolesc Health. 2018;2:582-590.