
 UK (English)
UK (English) Deutsch
Deutsch عربى
عربى Français
Français Hrvatska
Hrvatska Türkçe
Türkçe Română
Română
Что такое НЦЛ2?

Что такое НЦЛ2?
НЦЛ2 - генетическое заболевание.
это редкое генетическое заболевание, от которого страдают дети.1,2 Заболевание НЦЛ2:
- получило свое название в соответствии с наименованием гена CLN2/TPP1 , в котором при НЦЛ2 есть аномальная мутация2
- является одной из наиболее распространенных форм нейронального цероидного липофусциноза (НЦЛ)2
- НЦЛ –— это группа наследственных заболеваний, которые в совокупности именуются болезнью Баттена3
- Известный ранее под названием позднего инфантильного НЦЛ (потому что у большинства детей симптомы проявляются в возрасте от 2 до 4 лет),2
НЦЛ2 это генетическое заболевание, которое передается по наследству.
НЦЛ2 врожденное заболевание, хотя до появления симптомов иногда проходят месяцы и даже годы.2
аутосомно-рецессивное заболевание:2
- У каждого человека есть две копии гена НЦЛ2. У людей с НЦЛ2 в обоих унаследованных генах (по одному от каждого родителя) присутствуют мутации2
- У родителей ребенка с НЦЛ2 мутация есть в одном из генов НЦЛ22
- Родители являются носителями генетической мутации: это означает, что они здоровы, но могут передать мутацию своим детям2
- Если в генах НЦЛ2 у обоих родителей есть мутации, то существует2
- 25-процентная вероятность, что ребенок унаследует обе мутации, будет страдать НЦЛ2, и у него проявятс симптомы заболевания
- 50-процентная вероятность, что ребенок будет здоров, но будет носителем одного гена мутацияс мутацией НЦЛ2
- 25-процентная вероятность, что ребенок не станет носителем мутации НЦЛ2 и не заболеет.
Как ставится диагноз НЦЛ2?
Диагноз подтверждают с помощью генетического тестирования на предмет мутации в образце ДНК ребенка. Диагноз НЦЛ2 также можно подтвердить с помощью анализа фермента TPP1 (при НЦЛ2 обнарживается низкий уровень фермента).4
НЦЛ2 – вид лизосомальной болезни накопления, при которой поражаются клетки головного мозга5
Лизосомы присутствуют в каждой клетке. Лизосомы содержат ферменты, которые расщепляют и перерабатывают вещества в клетке. Один из этих ферментов –— TPP1.5
У детей с НЦЛ2 фермент TPP1 отсутствует или не функционирует должным образом. Когда этот фермент работает некорректно, в лизосомах клеток накапливаются определенные вещества (особенно в клетках головного мозга и глаз).3,6
При НЦЛ2 происходит накопление веществ внутри клеток мозга.3,6
.")
НЦЛ2 лизосомальная болезнь накопления (рисунок).
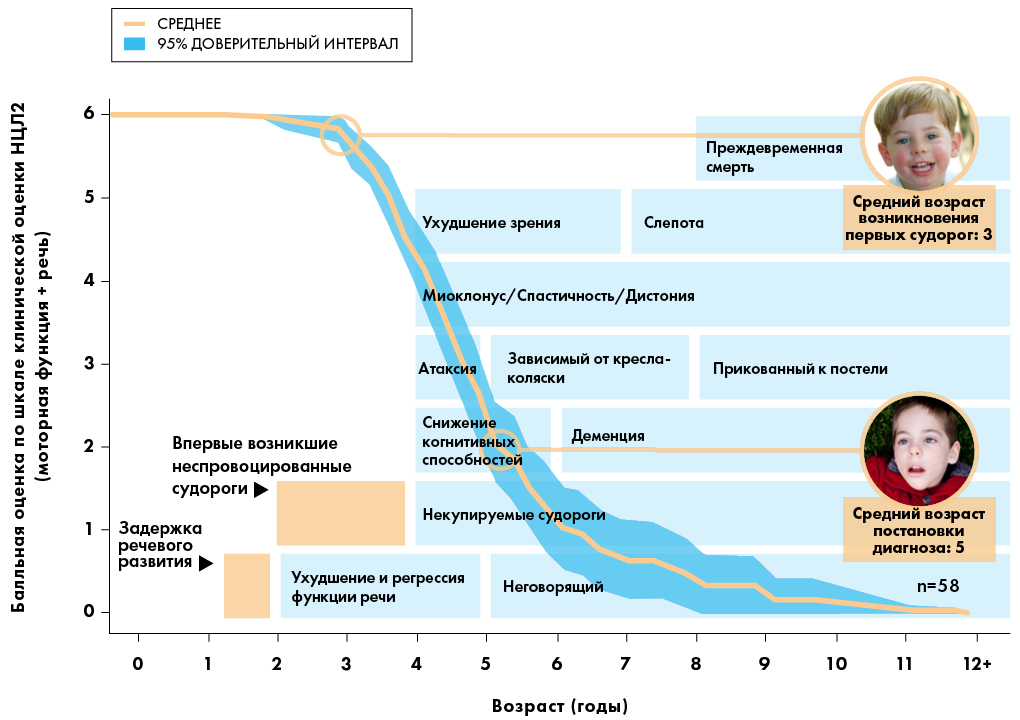
Со временем накопление приводит к повреждению клеток мозга и глаз, и эти органы перестают нормально функционировать. В этот период появляются симптомы НЦЛ2 (например, задержка речевого развития, судороги и нарушения зрения).1,3
Когда обычно появляются симптомы НЦЛ2?
Симптомы НЦЛ2 обычно со временем усиливаются, и включают:2,5,7-11
BDSRA logo
Основные источники
- Kohlschütter A, Schulz A. CLN2 disease (classic late infantile neuronal ceroid lipofuscinosis) [НЦЛ2 (классический поздний инфантильный нейрональный цероидный липофусциноз)]. Pediatr Endocrinol Rev. 2016;13 (Suppl 1):682-688.
- Mole SE, Williams RE. Neuronal ceroid-lipofuscinoses[Нейрональный цероидный липофусциноз]. 2001 Oct 10 [Updated 2013 Aug 1]. В: Pagon RA, et al., eds. GeneReviews®.
- Haltia M. The neuronal ceroid-lipofuscinoses: from past to present[Нейрональный цероидный липофусциноз: прошлое и настоящее.]. Biochim Biophys Acta. 2006;1762:850-856.
- Fietz M et al. Diagnosis of neuronal ceroid lipofuscinosis type 2 (CLN2 disease) [Диагностика нейронального цероидного липофусциноза 2 типа (НЦЛ2)]: Expert recommendations for early detection and laboratory diagnosis [Рекомендации экспертов по раннему выявлению и лабораторной диагностике]. Mol Genet Metab. 2016;119:160-167.
- Mole SE et al. Correlations between genotype, ultrastructural morphology and clinical phenotype in the neuronal ceroid lipofuscinoses [Корреляции между генотипом, ультраструктурной морфологией и клиническим фенотипом при нейрональном цероидном липофусцинозе]. Neurogenetics. 2005;6:107-126.
- Schulz A et al. NCL diseases–clinical perspectives [Заболевания НЦЛ: клинические перспективы]. Biochim Biophys Acta. 2013;1832:1801-1806.
- Chang M et al. CLN2 [НЦЛ2]. In: Mole S, Williams R, Goebel H,. The neuronal ceroid lipofuscinoses (Batten Disease) [Нейрональный цероидный липофусциноз (болезнь Баттена)]. 2nd ed. Oxford, United Kingdom: Oxford University Press; 2011:80-109.
- Pérez-Poyato MS et al. Late infantile neuronal ceroid lipofuscinosis: mutations in the CLN2 gene and clinical course in Spanish patients. [Поздняя инфантильная форма нейронального цероидного липофусциноза: мутации гена НЦЛ2 и клиническое течение у испанских пациентов.] . J Child Neurol. 2013;28:470-478.
- Worgall S et al. Treatment of late infantile neuronal ceroid lipofuscinosis by CNS administration of a serotype 2 adeno-associated virus expressing CLN2 cDNA [Лечение поздней инфантильной формы нейронального цероидного липофусциноза при введении в ЦНС аденоассоциированного вируса серотипа 2, экспрессирующего кДНК НЦЛ2]. Hum Gene Ther. 2008;19:463-474.
- Steinfeld R et al. Late infantile neuronal ceroid lipofuscinosis: quantitative description of the clinical course in patients with CLN2 mutations [Поздняя инфантильная форма нейронального цероидного липофусциноза: количественное описание клинического течения у пациентов с мутациями гена НЦЛ2]. Am J Med Genet. 2002;112:347-354.
- Schulz A et al. Neuronal ceroid lipofuscinosis-2 (CLN2) disorder, a type of Batten disease caused by TPP1 enzyme deficiency: current knowledge of the natural history from international experts [Нейрональный цероидный липофусциноз 2 типа (НЦЛ2) (болезнь Баттена), вызванный дефицитом фермента TPP1: естественное течение заболевания по актуальным данным международных экспертов]. Стендовая сессия, представленная на ежегодном симпозиуме SSIEM - Общество по изучению наследственных болезней обмена веществ (врожденных ошибок метаболизма). , сентябрь 2015 г.; Лион, Франция.